Contents
Background
"Normalization" refers to computational data transformations intended
to remove certain systematic biases from microarray data, such as dye
effects, intensity dependence, and spatial or print-tip effects. (In this
context, it doesn't necessarily have anything to do with the normal or
Gaussian distribution.) A wide variety of normalization approaches
have been proposed and employed in the literature. Each technique
relies on a set of assumptions about the ideal form of the data, and
attempt to make the data consistent with that ideal form by
computational manipulation. The database makes
available several different methods for "location" normalization:
total intensity normalization, M-A loess normalization, and
two-dimensional loess normalization. These techniques all adjust the
average of the data, either globally or stratified by intensity,
print-tip, and/or location. Scale normalization is also
available in conjunction with the loess methods; this adjusts the
range of the data.
This document briefly describes the normalization options available
within the database, and how to use them.
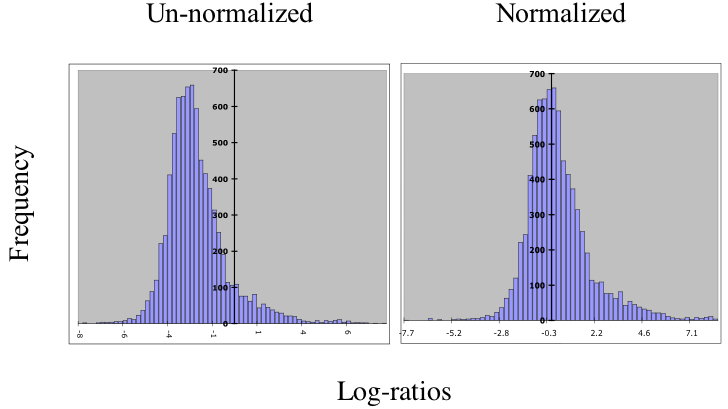
Default Total Intensity Normalization
Total intensity normalization relies on the assumption that most genes
do not respond to experimental conditions, and so the average log
ratio on the array should be zero.
Note that this may not be a
safe assumption for your data! A single, global, multiplicative
adjustment is performed so that the average log ratio is zero for well
measured spots. All spots are normalized using the same constant, regardless of
whether they were used in the calculation. In the database,
normalized channel 2 intensities are computed by dividing the raw
values by the normalization constant.
The normalization constant may be supplied by the user, or may be
calculated by the database's software. If calculated by the database,
the first step is to select good spots on which to base the
normalization. Two methods are available. Both begin by discarding flagged spots.
- The default "computed" normalization
procedure then selects non-flagged spots
for which the foreground intensity is well above background:
- If Scanalyze data : Both CH1GTB2 and CH2GTB2 (the fraction of the
pixels greater than the 1.5 times background of channels 1 and 2,
respectively) are greater than a threshold value.
- If GenePix or SpotReader data : Both % > B532+1SD and % > B635+1SD
(Percentage of spot pixels with intensities more than one standard
deviation above the background pixel intensity in channel 1 and 2)
are greater than a threshold value.
The threshold value is initially set to > 0.65. If fewer than 10% of
the spots in the
print pass these criteria, the program will use > 0.60. If fewer than
10% of the spots in the print pass the .60 threshold, the program will
use > 0.55. All spots that pass the 0.55 threshold are used in the
normalization calculation, regardless of how many there are. If more
than 10% of the spots pass any threshold, the program uses
those passing spots in the calculation and does not try a lower
threshold value.
- The "using regression correlation" method
selects non-flagged spots for which
the pixel-to-pixel regression correlation is > 0.6.
Complex Normalization Options
Several more complex normalization options are provided using the
Marray package for
BioConductor
(
Gentleman
et al., 2004), using the
R
statistical computing software.
Three location normalization options are provided:
- Median adjustment. This is essentially the
same as the database's default
total intensity normalization, but no spot filtering is performed.
Log ratios are adjusted globally such that the median log ratio is
zero; the database back-calculates normalized channel 2 intensities from the
normalized log-ratios.
- Intensity dependent normalization using local estimation. See
Yang
et al., 2002 and help documents on the BioConductor website for detailed
explanations of this approach. In essence, a smooth best-fit curve is
calculated for the dependence of log-ratio (M) on overall intensity
(A: log(base 2) of the geometric mean of the channel intensities).
Normalized log ratios are then given as the residuals from this curve
(and in the database, normalized channel 2 intensities are back-calculated from
the normalized log-ratios). Local estimation ("loess"), a regression
calculation weighted toward similar (in overall intensity value)
spots, is used to calculate the curve.
| Before Normalization |
After Global M-A Loess Normalization |
|---|
 |
 |
Intensity-dependent loess normalization
- Two-dimensional normalization using local estimation. The same
type of loess calculation is performed (see above references),
computing a smooth surface that gives the dependence of log-ratios on
spatial position across the microarray slide. Normalized log ratios
are given as residuals from this curve (essentially flattening the
surface) to eliminate spatial dependence in the data. In the database,
normalized channel 2 intensities are back-calculated from the
normalized log ratios. Spots are automatically stratified by
print-tip if you select this option (see below).
Loess calculations depend critically on the "span," a value between 0
and 1 that specifies the amount of data to include in each local
estimate, and thus the degree of smoothing. The value specified for
the span (default 0.4 in the database) will influence the results of loess
normalization, sometimes significantly. At the time of this writing
there are no generally-accepted methods for choosing an optimal span parameter.
The normalization calculation may be "stratified" by print-tip
(sector). This will cause the normalization to be performed
separately on each sector. This is generally appropriate for
pin-printed microarrays, in which print-tip effects are common;
stratification by print-tip will eliminate much of the print-tip
effect on the data. In the database, if you select two-dimensional
normalization (above), spots will automatically be stratified by
print-tip. Stratification is not available for the default
normalization - use the marray median adjustment, instead.
"Scale" normalization adjusts the range of data, rather than the
center of the distribution. This makes data more comparable across
arrays, by eliminating differences in the range of response to
conditions. Of course, this may not be appropriate; it is generally
advised only when the absolute scale of response is not relevant (or
not well measured). The database supports division of all values by
the median absolute deviation (MAD) of the array (or sector if
print-tip stratification is selected). This may be combined with
location normalization (median adjustment, intensity-dependent loess,
or 2-D loess functions only), in which case scale normalization will
be performed following location normalization.
How to renormalize experiments
Arrays may be renormalized one at a time by following the "Select
normalization options" link while editing the experiment. To
renormalize all arrays in an
arraylist,
follow the
Batch Renormalize Data link in the list of all programs.
Only GenePix,
ScanAlyze, and SpotReader data may be renormalized within the database.
Agilent and Affymetrix software provide other options for
normalization prior to loading into the database.
