
PUMAdb : Help Entering Results from Experiments
Your session is inactive. Login
Contents
- Links to More Specific Help Pages
- Description
- Types of Microarray data accepted
- First Time Users
- Entering Your Data
- Data Files Needed to Load Your Data
- Location of data files
The Experiment and Result Entry form is the starting point from which to load microarray results into the database. To do this, you need an "unrestricted" account and an account on loader.princeton.edu. For more information regarding both the accounts and access required to enter results into the database, please refer to the PUMAdb Accounts and Access document.
| Software | Version |
|---|---|
| Agilent | A.6.1.1, A.7.1.1 and A.7.1.2 |
| Affymetrix | MAS 5 (5.0, 5.1), GCOS (1.0, 1.1), and dChip 1.3 |
| GenePix | 3.0, 4.1, and 5.0 |
| NimbleScan | 2.1.16 |
| ScanAlyze | All |
| SpotReader | 1.0 |
| Affymetrix Tiling | SNPScanner |
For all types of feature extraction software, you need the following things before you enter an experiment into the database. The table below describes these items. All files submitted can be compressed or uncompressed. Compressed files have the normal suffix with .gz on the end; an example would be .dat.gz .
| Item | Unique | Required | Max # of characters | Notes |
|---|---|---|---|---|
| Print name1 | * | N/A | often the spotlist or Godlist name | |
| Slide name | *2 | * | 100 | Usually a systematic name assigned by the slide printing facility |
| Experiment Name | *2 | * | 100 | |
| Green3 or Single4 Channel Description | * | 100 | ||
| Red Channel Description3 | * | 100 | ||
| Experiment Description | 2000 | Unformatted text to describe experiment details | ||
| Category5 | * | 30 | Choose from a list in the database | |
| Subcategory5 | * | 30 | Choose from a list in the database | |
| Experiment Type6 | * | 40 | Choose from a list in the database | |
| Normalization Type7 | * | 30 | Choose from a list in the database | |
| Norm Value7 | * | 30 | For normalization type "user-defined", a normalization value must be entered. | |
| Result Set Name8 | * | * | 100 | The name of your result set |
| Result Set Description8 | 240 | Free text description of your result set | ||
| Probe Set Algorithm4 | * | 240 | Accepted values are: 'Affymetrix MAS 5', 'dChip MBEIs', or 'SNPScanner' (tiling only), depending on what software was used to 'normalize' your data. |
Table Notes1 If you don't know which print to use for your experiment, after login, click Print List under "List Data" from the Main page or click "Print Name" on the individual experiment entry form. For further information, contact the microarray database curators.
2 Every slide name (e.g., array serial #) and experiment (hybridization) name must be unique - you may not re-use them, or use the same names as any other user. Result set names may be re-used, but only once per slide - you may have a result set called "simple normalization" for each of your slides, but only one per slide.
3 This field is required only for GenePix, ScanAlyze, or Agilent data.4 This field is required only for Affymetrix data.
5 The database requires a category and subcategory for each experiment, both of which are chosen from lists stored in the database. Any category can be paired with any subcategory to describe an experiment. Categories, subcategories, and their descriptions can be found by clicking Category or SubCategory under "List Data" on the Main page, or by clicking Experiment Category or Experiment SubCategory on the individual experiment entry form. If you need a category or subcategory which is not already in the database, contact the microarray database curators.6 This field is required for NimbleGen data, and is ignored for the others. Experiment types and their descriptions can be found by clicking Experiment types under "List Data" on the Main page. If you need an experiment type which is not already in the database, please send an email to the microarray database curators.
7 This field is always required, but it is ignored for Agilent and Affymetrix data.
8 These values are needed for Affymetrix and Agilent data only. See the note on unique slide, experiment, and result set names above2.
| Item | Suffix | Max # of characters | Notes |
|---|---|---|---|
| Data File1 | .dat, .gpr, .srr, .txt | 250 | Please check
the print dimensions within
the database before gridding your array.2 For Agilent data, be sure that you have the text file and not the xml file. |
| Grid File | .sag, .gps, .sra, .shp | 250 | |
| Green Scan File3 | .tif | 250 | Typically the 532nm scan, if the original image was split. If the image was not split, you will be prompted only for the single file name. |
| Red Scan File3 | .tif | 250 | Typically the 635nm scan, if the original image was split. If the image was split, this will not be an option. |
Table Notes1 Do not change the default column names in the data file. SpotReader in particular gives you a choice of "channel shortcut names." Any of the default two-channel options are acceptable (Ch1/Ch2, Cy3/Cy5, Green/Red, 532/635). Any other names will cause experiment loading to fail.
2 Attempts to load array data not matching print dimensions (tips/blocks x rows x columns) are disallowed. If using GenePix, do not pre-filter any of the spot-features. Only a full gpr file, with an entry for every spot, in order, can be loaded.
3 Automatic .gif generation requires that you submit either one or two .tif (not .scn) files when entering your experiment (one for each channel, if submitting two). The automatic .gif generation fails occasionally. However, if your .gif is not created at experiment entry, the microarray database curators can make it for you in most cases. It is also possible to upload a preferred .gif file if you don't like the generated ones.
| Item | Suffix | Max # of characters | Notes |
|---|---|---|---|
| Data File | .dat | 250 | The image file is generated by the Affymetrix chip scanning software. It is a 16-bit tiff file and will be archived and converted to a 8-bit gif file for viewing in the database. This file is optional. |
| Cell File | .cel | 250 | This file is generated from the .dat file by the Affymetrix MAS 5 or Affymetrix GeneChip Operating Software (GCOS). The native .CEL file format for GCOS is a proprietary binary format. To upload GCOS .CEL files into the database, open the GCOS Manager program and export the .CEL file. This converts it into a text file our software can understand. |
| Gene File | .txt, .xls | 250 | This file is generated from the .cel file by Affymetrix MAS 51, Affymetrix GeneChip Operating Software (GCOS)2, and dChip3. To upload the Probe Set file into the database it needs to be exported from the analysis software as a tab-delimited text file, see Preparing a probe set file below. |
| Experiment File | .exp | 250 | This file is generated by the Affymetrix chip scanning software and contains chip protocol information. |
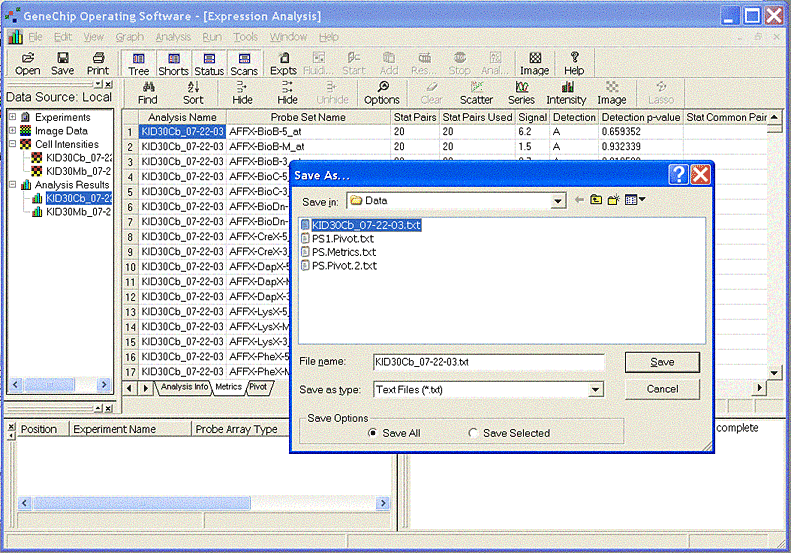
1Affymetrix MAS 5 Open the Probe Set intensity file (.CHP file) for a single chip. Select either the 'Pivot' tab or the 'Metrics' tab and save as a text file using the menu File -> Save as.
Enter 'Affymetrix MAS 5' for Probe Set Algorithm when loading the experiment.
2 Affymetrix GeneChip Operating Software (GCOS)
Open the Probe Set intensity file (.CHP file) for a single chip.
Select either the 'Pivot' tab or the 'Metrics' tab and
save as a text file using the menu File -> Save as.
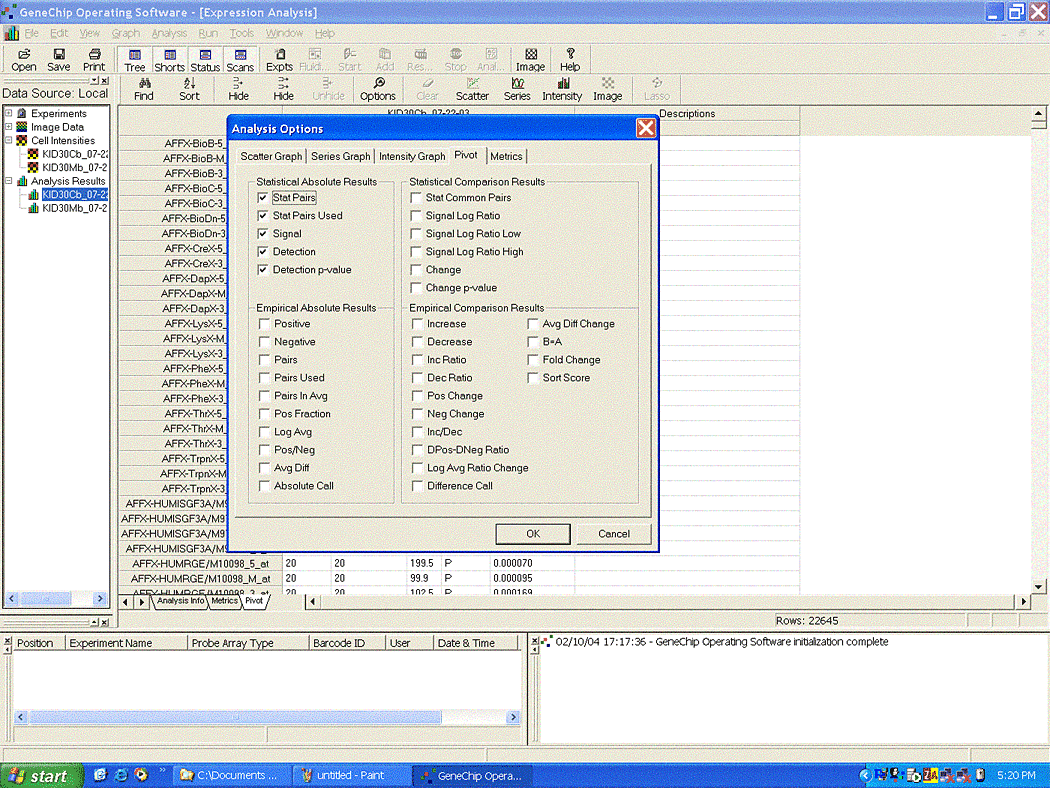
If saving from the 'Pivot' tab, select all of the 'Statistical
Absolute Result' columns from the menu Analyis -> Options -> Pivot tab.
Enter 'Affymetrix MAS 5' for Probe Set Algorithm when loading the experiment.
3dChip
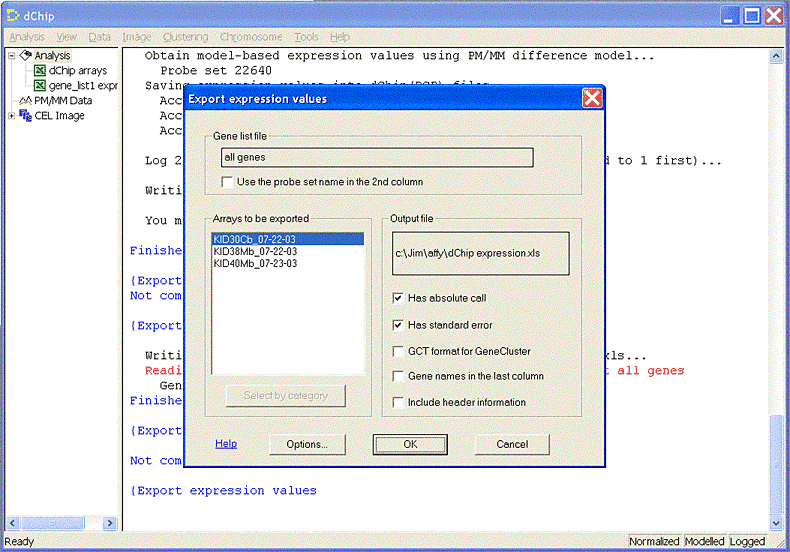
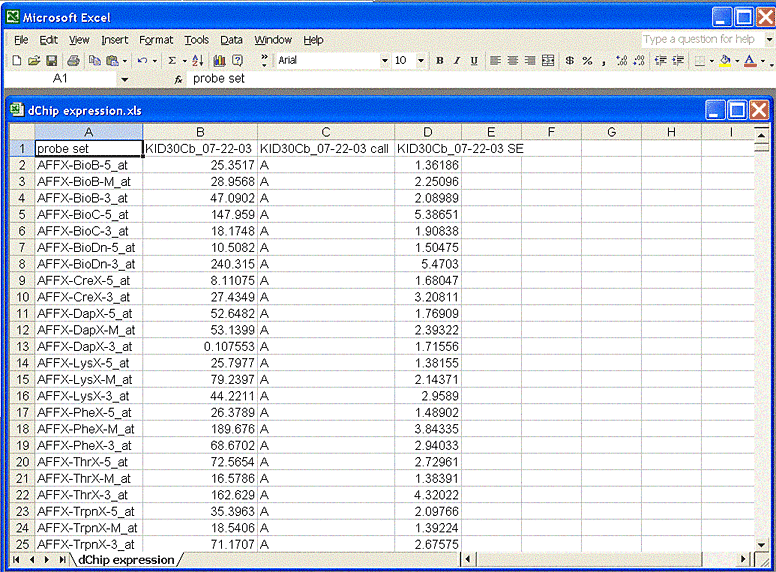
Open one or more chips. Select menu Tools -> Export Data. Then select a single chip, an export file name, and absolute call and standard error columns to export the Probe Set intensity values. An example exported file is shown here. If you open it in Excel, remember to save it as tab-delimited text.
Enter 'dChip MBEIs' for Probe Set Algorithm when loading the
experiment.
Table Notes (Preparing a Probe Set File)
Affymetrix Tiling (SNPScanner)
| Item | Suffix | Max # of characters | Notes |
|---|---|---|---|
| Data File | .dat | 250 | The image file is generated by the Affymetrix chip scanning software. It is a 16-bit tiff file and will be archived and converted to a 8-bit gif file for viewing in the database. |
| Cell File | .cel | 250 | This file is generated from the .dat file by Affymetrix Software. |
| Results File | .txt | 250 | This file is generated by SNPScanner from the .cel file. |
| Experiment File | .exp | 250 | This file is generated by the Affymetrix chip scanning software and contains chip protocol information. |
| Item | Suffix | Max # of characters | Notes |
|---|---|---|---|
| Image File | .tif | 250 | The image file should be a tiff file of the single channel scan. This file will be archived and a gif copy of it will be created for viewing in the database. |
| Cell File | .xys | 250 | This file contains processed result data for each feature on the slide. It is a tab-delimited text file. |
| FTR File | .ftr | 250 | This file is a tab-delimited text file that contains result information about features on the slides. |
| Gene Intensity File | .txt, .calls | 250 | This file is a tab-delimited text file that contains result information for genes. |
Data can be loaded by first placing files into either :
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}